Bioinformatics Pipeline Engineering: Do This Not That: Split Tasks
Overview
In the last installment of this series, we talked about how to handle large files without bringing down the wrath of your local sys admin. Today we're tackling how to think about breaking up tasks.
You've probably written it. I've definitely written it. It looks perfectly reasonable when you first start:
for sample in samples:
run_qc(sample)
align(sample)
call_variants(sample)
(I am aware that it is unlikely you'd write this exact code. Suspend your disbelief and walk with me. We're going somewhere with this.)
Simple. Readable. Gets the job done.
Until it doesn't!
Maybe you're now running a 200GB alignment step on the same node where you're doing QC, or you can't checkpoint a failed run, or your HPC scheduler has no idea how to allocate resources for a step that sometimes needs 4GB and sometimes needs 400GB. Maybe your local admin is upset because nodes are underutilized and she's got annoyed scientists waiting for a machine. (Day in the life!)
In this post we'll look at splitting monolithic tasks into discrete CLI commands, and then show how naturally those map to a Nextflow pipeline.
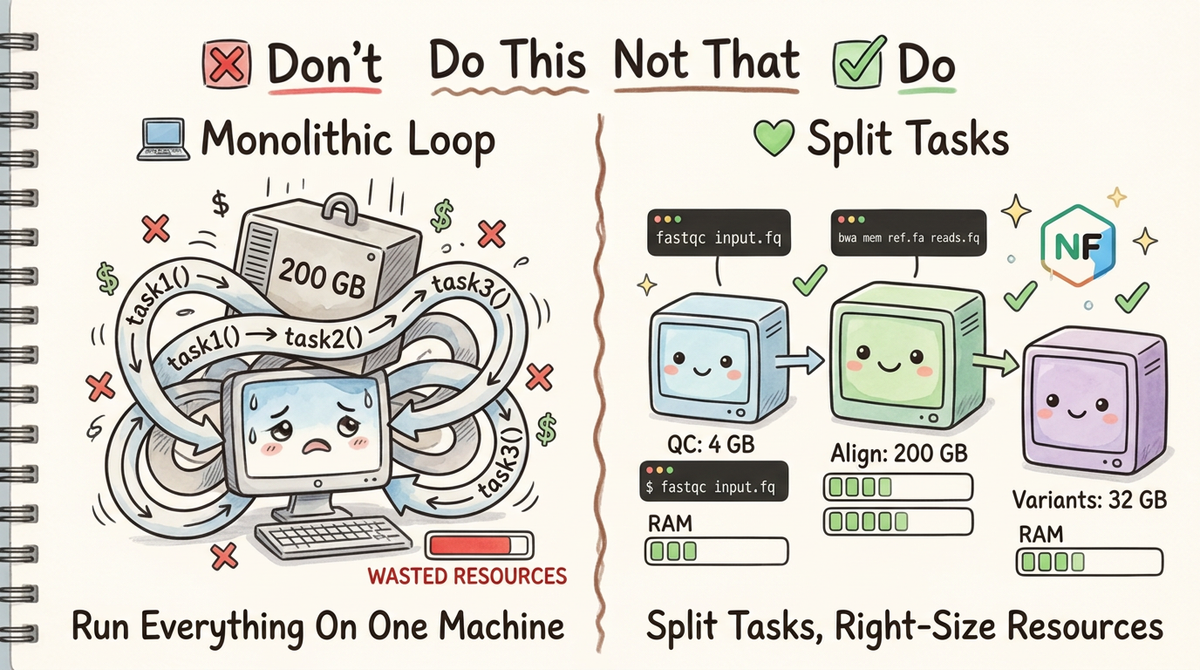
Don't: Lump Everything Into a Single Script Loop
The Problem
Here's a pattern that feels intuitive but causes real pain at scale:
# ❌ DON'T DO THIS
def run_pipeline(samples):
for sample in samples:
task1(sample) # QC: uses ~4 GB RAM
task2(sample) # Alignment: uses ~200 GB RAM
task3(sample) # Variant calling: uses ~32 GB RAM
When every task is tangled together in a loop, you have no control. The whole pipeline runs on a single machine, allocating resources for the worst-case step across every step. You can't restart from the middle if something fails. Your HPC scheduler can't optimize placement. And three months from now, when a colleague asks "what does task2 actually do?", the answer lives somewhere inside a 400-line script.
Why This Hurts in Practice
Consider a real-world scenario: you have 50 samples, QC takes 4 GB, alignment takes 200 GB, and variant calling takes 32 GB.
(Of course you would just use an existing pipeline for this workflow. Pretend we have some custom barcodes or something.)
With the monolithic approach:
- Every step runs on a node provisioned for the largest step (200 GB)
- You're paying for 200 GB of RAM during QC, when you only need 4 GB
- A single failure at sample 49 means restarting everything from scratch
- Adding a new step means modifying the loop and hoping you don't break the others
- There's no obvious place to document what each step does or what it expects
This is exactly the kind of thing that quietly costs teams thousands of dollars in cloud compute every month.
Do: Split Tasks into Discrete CLI Commands
Step 1 — Give Each Task Its Own Typer Command
Typer is a fantastic library for building CLIs from Python functions. The key insight is that a CLI command is documentation: the function signature, types, and help strings tell you exactly what goes in and what comes out.
Here's what the refactored version looks like:
# ✅ DO THIS
import typer
from pathlib import Path
app = typer.Typer()
@app.command()
def qc(
input_fastq: Path = typer.Argument(..., help="Path to input FASTQ file"),
output_dir: Path = typer.Argument(..., help="Directory to write QC results"),
min_quality: int = typer.Option(20, help="Minimum base quality score"),
):
"""
Run quality control on a FASTQ file.
Expected resource usage: ~4 GB RAM, 2 CPUs.
"""
output_dir.mkdir(parents=True, exist_ok=True)
# ... QC logic here ...
typer.echo(f"QC complete. Results written to {output_dir}")
@app.command()
def align(
input_fastq: Path = typer.Argument(..., help="Path to QC-passed FASTQ file"),
reference: Path = typer.Argument(..., help="Path to reference genome"),
output_bam: Path = typer.Argument(..., help="Path to output BAM file"),
threads: int = typer.Option(16, help="Number of alignment threads"),
):
"""
Align reads to a reference genome using BWA-MEM2.
Expected resource usage: ~200 GB RAM, 16 CPUs.
"""
output_bam.parent.mkdir(parents=True, exist_ok=True)
# ... alignment logic here ...
typer.echo(f"Alignment complete. BAM written to {output_bam}")
@app.command()
def call_variants(
input_bam: Path = typer.Argument(..., help="Path to aligned BAM file"),
reference: Path = typer.Argument(..., help="Path to reference genome"),
output_vcf: Path = typer.Argument(..., help="Path to output VCF file"),
min_depth: int = typer.Option(10, help="Minimum read depth to call a variant"),
):
"""
Call variants from an aligned BAM file using GATK HaplotypeCaller.
Expected resource usage: ~32 GB RAM, 4 CPUs.
"""
output_vcf.parent.mkdir(parents=True, exist_ok=True)
# ... variant calling logic here ...
typer.echo(f"Variant calling complete. VCF written to {output_vcf}")
if __name__ == "__main__":
app()
Now each task is invocable on its own:
# Run just QC
python pipeline.py qc sample1.fastq.gz ./results/qc/
# Run just alignment
python pipeline.py align results/qc/sample1.fastq.gz ref/hg38.fa results/bam/sample1.bam
# Run just variant calling
python pipeline.py call-variants results/bam/sample1.bam ref/hg38.fa results/vcf/sample1.vcf.gz
And you get auto-generated help for free:
$ python pipeline.py align --help
Usage: pipeline.py align [OPTIONS] INPUT_FASTQ REFERENCE OUTPUT_BAM
Align reads to a reference genome using BWA-MEM2.
Expected resource usage: ~200 GB RAM, 16 CPUs.
Arguments:
INPUT_FASTQ Path to QC-passed FASTQ file [required]
REFERENCE Path to reference genome [required]
OUTPUT_BAM Path to output BAM file [required]
Options:
--threads INTEGER Number of alignment threads [default: 16]
--help Show this message and exit.
That's documentation that stays in sync with the code, always.
Step 2 — Wrap Commands in a Container
Once your tasks are discrete CLI commands, containerizing them is straightforward. Each step can have its own Docker image with exactly the dependencies it needs—no more shipping a 15 GB image because alignment and QC happen to live in the same script.
# Dockerfile.align
FROM ubuntu:22.04
RUN apt-get update && apt-get install -y bwa-mem2 samtools python3-pip
RUN pip install typer
COPY pipeline.py /opt/pipeline.py
ENTRYPOINT ["python", "/opt/pipeline.py", "align"]
Build and run:
docker build -f Dockerfile.align -t my-pipeline/align:latest .
# or singularity run!
docker run --rm \
-v /data:/data \
my-pipeline/align:latest \
/data/sample1.fastq.gz /data/hg38.fa /data/results/sample1.bam
Step 3 — Drop Them Into Nextflow
Now that each task is a standalone command with well-defined inputs and outputs, translating to Nextflow is almost mechanical. Each CLI command becomes a Nextflow process. Once we have our code in a workflow manager we get all kinds of sanity checks! We can write checks, resume, look at memory consumption, etc.
The Nextflow Module
// modules/qc.nf
process QC {
tag "$sample_id"
label 'process_low' // 4 GB RAM, 2 CPUs
container 'my-pipeline/qc:latest'
input:
tuple val(sample_id), path(fastq)
output:
tuple val(sample_id), path("${sample_id}_qc/"), emit: qc_results
script:
"""
python pipeline.py qc \\
${fastq} \\
${sample_id}_qc/
"""
}
// modules/align.nf
process ALIGN {
tag "$sample_id"
label 'process_high_memory' // 200 GB RAM, 16 CPUs
container 'my-pipeline/align:latest'
input:
tuple val(sample_id), path(fastq)
path reference
output:
tuple val(sample_id), path("${sample_id}.bam"), emit: bam
script:
"""
python pipeline.py align \\
${fastq} \\
${reference} \\
${sample_id}.bam \\
--threads ${task.cpus}
"""
}
// modules/call_variants.nf
process CALL_VARIANTS {
tag "$sample_id"
label 'process_medium' // 32 GB RAM, 4 CPUs
container 'my-pipeline/call-variants:latest'
input:
tuple val(sample_id), path(bam)
path reference
output:
tuple val(sample_id), path("${sample_id}.vcf.gz"), emit: vcf
script:
"""
python pipeline.py call-variants \\
${bam} \\
${reference} \\
${sample_id}.vcf.gz
"""
}
The Workflow
// main.nf
include { QC } from './modules/qc'
include { ALIGN } from './modules/align'
include { CALL_VARIANTS } from './modules/call_variants'
workflow {
samples_ch = Channel
.fromPath(params.samplesheet)
.splitCsv(header: true)
.map { row -> tuple(row.sample_id, file(row.fastq)) }
reference_ch = file(params.reference)
QC(samples_ch)
ALIGN(QC.out.qc_results, reference_ch)
CALL_VARIANTS(ALIGN.out.bam, reference_ch)
}
And the resource labels map to concrete allocations in nextflow.config:
// nextflow.config
process {
withLabel: 'process_low' {
cpus = 2
memory = '4 GB'
}
withLabel: 'process_medium' {
cpus = 4
memory = '32 GB'
}
withLabel: 'process_high_memory' {
cpus = 16
memory = '200 GB'
}
}
Now Nextflow knows exactly how much memory each step needs, and your scheduler—whether that's AWS Batch, SLURM, or Google Cloud Life Sciences—can place each process on an appropriately sized node.
Once you have your pipeline in Nextflow you no longer have to worry about orchestrating your pipeline. Nextflow will take over on assigning resources whether you're working on a traditional HPC, AWS Batch, or locally. You'll automatically get failover and retries. Each steps success/failure, time, and resource management will be logged automatically.
Why This Matters: The Three Benefits
1. Organization
A Typer command is self-documenting by design. The function name becomes the command name. Type annotations become argument validation. Docstrings become --help output. When you (or a new team member) comes back to this code in six months, every step has an obvious entry point, clear inputs and outputs, and inline documentation. No more hunting through a 500-line script to figure out what step 3 actually does.
2. Resource Consumption
This is where splitting tasks pays real dividends. When QC needs 4 GB and alignment needs 200 GB, running them in the same process means every sample occupies a 200 GB node for the entire pipeline—even during QC. With discrete Nextflow processes, each step runs on the smallest node that can handle it:
| Step | RAM Required | Node Cost (est.) |
|---|---|---|
| QC | 4 GB | $0.02/hr |
| Alignment | 200 GB | $0.85/hr |
| Variant Calling | 32 GB | $0.14/hr |
Running monolithically at the highest requirement for 50 samples means paying for 200 GB throughout. Splitting tasks means you only pay for what each step actually needs. At scale, that difference is not small.
3. Pipeline Integration
The moment each task is a discrete CLI command, it becomes a composable unit. Nextflow (or Snakemake, or WDL) can:
- Parallelize across samples automatically—no manual multiprocessing
- Checkpoint and resume from any step without rerunning the whole pipeline
- Retry failed steps with different resource allocations
- Cache completed steps so re-runs skip work that's already done
- Scale from your laptop to 1,000-node HPC clusters without changing a line of Python
The CLI is the interface. The pipeline orchestrator is the caller. They don't need to know anything about each other.
Best Practices Checklist
- ✅ One command per logical step: If you find yourself putting two
typer.Options that only apply to half the function, you probably have two commands - ✅ Document resource expectations in docstrings: Future you will thank you
- ✅ Use
Pathtypes: Typer validates existence for you; no moreFileNotFoundErrorburied in the middle of a long run - ✅ Match CLI arguments to Nextflow inputs: Keep the interface consistent so the Nextflow script is a thin wrapper, not a translation layer
- ✅ Test each command independently: If a step can't run standalone, it can't run reliably in a pipeline
Key Takeaway
The monolithic loop is a natural starting point, but it creates a ceiling. Splitting your pipeline into discrete CLI commands—each with defined inputs, outputs, and resource requirements—removes that ceiling entirely.
Split your tasks. Document your interfaces. Let the scheduler worry about the rest.
Next in the "Do This Not That" series: Batched vectorized operations over multiprocessing