Bioinformatics Pipeline Engineering: Do This Not That: Large Files
Overview
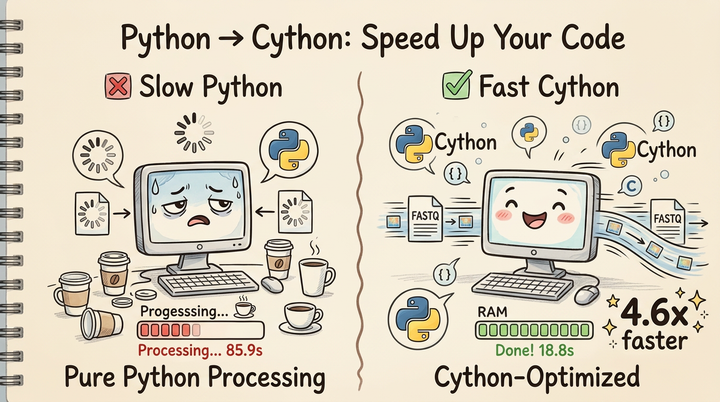
If you've worked with bioinformatics data, you've likely encountered the dreaded experience: your Python script chugging along, consuming memory at an alarming rate, and then—crash. Out of memory. Your computer becomes unresponsive, and you're left frustrated wondering why your beautiful analysis script decided to obliterate your system. Maybe you've even taken down an entire HPC!
Here's the thing: working with large bioinformatics files—FASTQ sequences, whole genome VCF files, BAM alignments, protein structure files—requires a fundamentally different approach than analyzing a CSV with a few thousand rows. These files often contain millions or billions of records, and the naive approach of "just load it all into memory" is often a recipe for a headache.
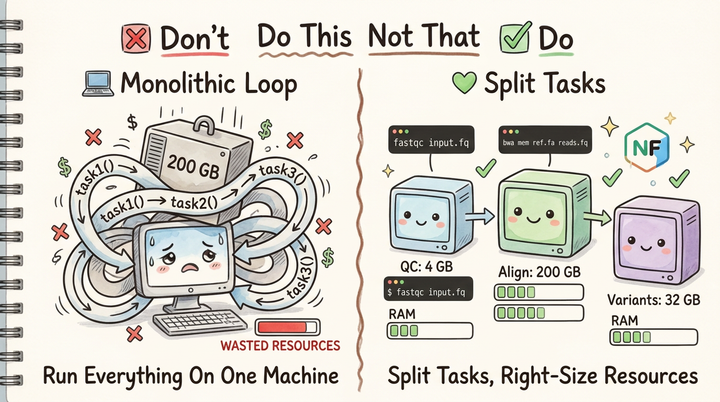
In this "Do This Not That" series, we're going to explore practical patterns for optimizing bioinformatics software. Today, we're tackling file handling at scale.
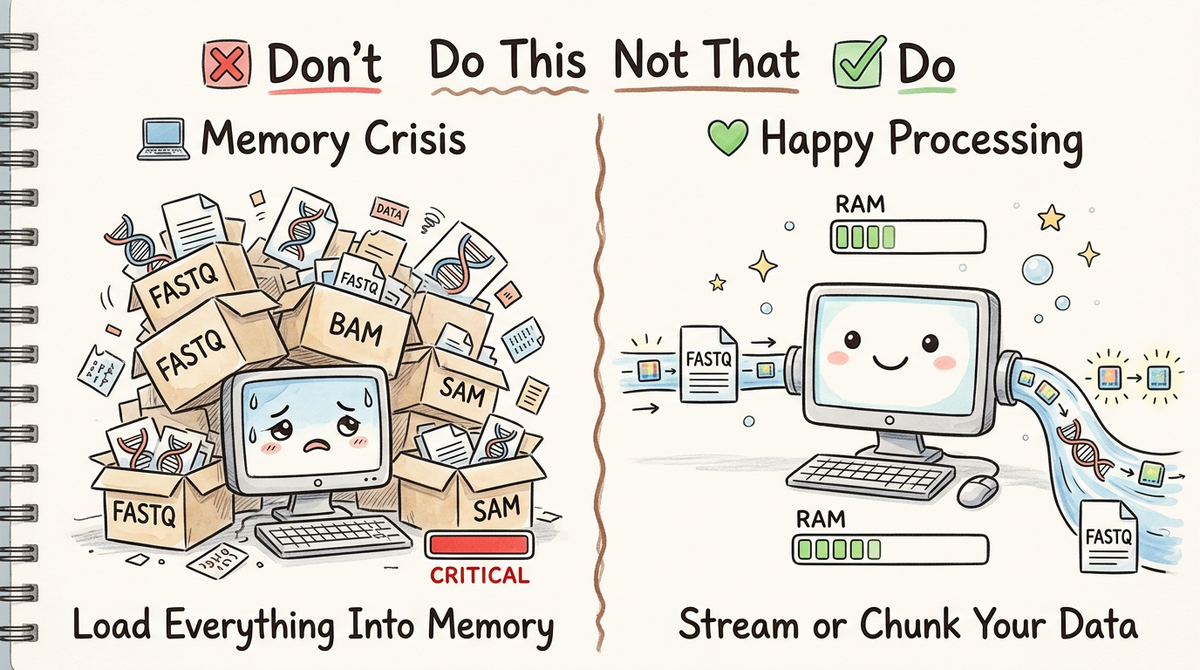
Don't: Load Everything Into Memory
The Problem
The worst approach—and unfortunately the most common among newcomers—is to load an entire large file into memory:
# ❌ DON'T DO THIS
def analyze_fastq(filepath):
"""Load entire FASTQ file into memory. Catastrophic for large files."""
sequences = []
with open(filepath, 'r') as f:
lines = f.readlines() # Load ALL lines into memory
# Parse fastq into records
for i in range(0, len(lines), 4):
header = lines[i].strip()
seq = lines[i+1].strip()
plus = lines[i+2].strip()
qual = lines[i+3].strip()
sequences.append({
'header': header,
'sequence': seq,
'quality': qual
})
return sequences
# This will crash with a 50GB FASTQ file
results = analyze_fastq("huge_genomic_dataset.fastq")
Why this fails:
- Memory explosion: A 100GB FASTQ file needs 100GB+ of RAM just to store the raw text, then additional memory for parsing into data structures
- Python object overhead: Each string and dictionary adds metadata overhead (Python objects are not memory-efficient)
- No streaming: You're waiting for the entire file to load before processing even one record
- Swap thrashing: When RAM is exceeded, your OS swaps to disk, slowing everything to a crawl
Real-World Impact
On a machine with 32GB of RAM, trying to load a 200GB human genome VCF file will:
- Use up all available RAM
- Trigger swap, using disk as virtual memory (100x slower)
- Freeze your system
- Eventually crash with an out-of-memory error
You've now lost all time and resources with nothing to show for it. You may be getting annoyed emails from your local sys admin.
Do: Stream or Chunk Your Data
Approach 1: Streaming (Process Line-by-Line)
The most memory-efficient approach is to process data line-by-line or record-by-record without storing everything:
# ✅ DO THIS - Streaming approach
def analyze_fastq_stream(filepath):
"""Process FASTQ records one at a time."""
def fastq_reader(file_obj):
"""Generator: yield one FASTQ record at a time."""
while True:
header = file_obj.readline().strip()
if not header:
break
seq = file_obj.readline().strip()
plus = file_obj.readline().strip()
qual = file_obj.readline().strip()
yield [header, seq, qual]
# Process records as they're read (memory-constant)
stats = {
'total_records': 0,
'avg_length': 0,
'total_bases': 0,
}
with open(filepath, 'r') as f:
for record in fastq_reader(f):
# Process each record individually
stats['total_records'] += 1
stats['total_bases'] += len(record[1])
stats['avg_length'] = stats['total_bases'] / max(stats['total_records'], 1)
return stats
# This handles terabyte-scale files with constant memory
results = analyze_fastq_stream("huge_genomic_dataset.fastq")
Advantages:
- Constant memory: Only one record in memory at any time
- Immediate processing: Start analyzing data before the file finishes loading
- Infinite scalability: Handle files larger than your system's RAM
- Graceful degradation: Works whether your file is 1MB or 1TB
Approach 2: Chunked Reading (Process in Batches)
For operations that benefit from batch processing (like machine learning inference), read in managed chunks.
Typically, when I do this, its because I want to interim results to parquet files. We get one parquet file per chunk, which can then be spread out across a scheduler.
import pyarrow as pa
import pyarrow.parquet as pq
# ✅ DO THIS - Chunked approach
def analyze_fastq_chunked(filepath, chunk_size=10000):
"""Process FASTQ records in fixed-size batches."""
def batch_fastq_reader(file_obj, batch_size):
"""Generator: yield batches of FASTQ records."""
seqs = []
headers = []
quals = []
while True:
header = file_obj.readline().strip()
if not header:
if batch:
yield batch
break
seq = file_obj.readline().strip()
plus = file_obj.readline().strip()
qual = file_obj.readline().strip()
# batch.append([header, seq, qual])
headers.append(header)
seqs.append(seq)
quals.append(qual)
if len(batch) >= batch_size:
yield headers, seqs, quals
headers = []
seqs = []
quals = []
# Process in batches (bounded memory usage)
batch_num = 0
with open(filepath, 'r') as f:
for batch in batch_fastq_reader(f, chunk_size):
# Process this batch (e.g., run ML inference)
headers, seqs, quals = process_sequences_batch(batch)
table = pa.Table.from_pydict({'header': headers, 'sequence', : seqs, 'quality': quals})
# save additional processing for later
pq.write_table(table, f'fastq-records-{batch_num}.parquet')
print(f"Processed batch of {len(batch)} records")
batch_num += 1
return all_results
Once you have your data in an arrow/pandas table, you can replace many bioinformatics string mantching type functions. Running code against all elements in the column of a table is nearly always faster than running code against all items in a list. (We'll be talking about this next!)
Advantages:
- Bounded memory: Memory usage stays constant regardless of file size
- Batch efficiency: Some operations (ML, vectorized computing) are faster in batches
- Progress tracking: You can report progress as batches complete
- Parallelizable: Each batch can be processed independently
- Simple: Keep your code as simple as possible. When running code millions of times even small delays really add up!
Approach 3: Using Libraries Built for Large Files
This one is a bit fuzzy. I'm all for using code that I didn't write. Ok? All about it.
But my dreams just don't always match my reality of needing my code to run as quickly as possible. AWS credits don't grow on trees!
Bioinformatics libraries are built to parse bioinformatics data. This can be a good thing. Some libraries, such as pysam, are optimized and fast.
Other libraries, such as BioPython, less so. Sometimes you need to do what you need to do, but most often you'll be better off rolling your own, or better yet using a library such as pyarrow.
# ✅ SOMETIMES DO THIS - Use specialized libraries
from Bio import SeqIO # BioPython
# BioPython: Built-in streaming parser
def analyze_with_biopython(fastq_file):
"""BioPython handles streaming automatically."""
stats = {'count': 0, 'gc_content': 0}
for record in SeqIO.parse(fastq_file, "fastq"):
# Process each record without loading file into memory
stats['count'] += 1
stats['gc_content'] += record.seq.count('G') + record.seq.count('C')
return stats
For even faster batch operations on accumulated sequences, use PyArrow's vectorized string functions:
# ✅ BEST FOR BATCH PROCESSING: Accumulate and use PyArrow compute functions
import pyarrow as pa
import pyarrow.compute as pc
def analyze_with_pyarrow(fastq_file):
"""Accumulate sequences in batches, use vectorized string counting."""
sequences = []
with pysam.FastqFile(fastq_file) as fq:
for record in fq:
sequences.append(record.sequence)
# Process in batches of 10,000
if len(sequences) >= 10000:
# Use PyArrow's vectorized count_substring for speed
seq_array = pa.array(sequences)
total_gc = int(pc.count_substring(seq_array, pattern="G")) + int(pc.count_substring(seq_array, pattern="C"))
print(f"Batch GC content: {total_gc}")
sequences = []
# Handle remaining sequences
if sequences:
seq_array = pa.array(sequences)
total_gc = int(pc.count_substring(seq_array, pattern="G")) + int(pc.count_substring(seq_array, pattern="C"))
print(f"Final batch GC content: {total_gc}")
PySam is nearly always going to be fastest for file I/O. It accepts most major bioinformatics file format types.
# ✅ SOMETIMES DO THIS - Use specialized libraries
# PysSAM: Efficient BAM file iteration
import pysam # SAM/BAM handling
def analyze_bam_stream(bam_file):
"""PysSAM streams BAM records efficiently."""
stats = {'total_reads': 0, 'mapped_reads': 0}
with pysam.AlignmentFile(bam_file, "rb") as bam:
for read in bam:
stats['total_reads'] += 1
if not read.is_unmapped:
stats['mapped_reads'] += 1
return stats
Why this matters:
- Battle-tested: These libraries are optimized for bioinformatics use cases
- Zero-copy operations: Many use C/C++ backends that don't allocate excess memory
- Standard patterns: Your colleagues will recognize the code
- Feature-rich: Built-in support for filtering, transforming, etc.
Compressed Files: Even More Important
Large bioinformatics files are almost always compressed (gzip, bgzip, zstd). Reading compressed files requires special attention:
# ❌ DON'T: Decompress entire file first
import gzip
import shutil
# This creates a huge temporary uncompressed file
with gzip.open('huge_file.fastq.gz', 'rb') as f_in:
with open('huge_file.fastq', 'wb') as f_out:
shutil.copyfileobj(f_in, f_out) # Now you have 200GB on disk
# ✅ DO: Use smartopen for transparent decompression
from smart_open import open as smart_open
def stream_compressed_fastq(compressed_file):
"""Read compressed file without decompressing to disk.
smartopen automatically detects compression format (gzip, bzip2, zstd, etc.)
and handles decompression transparently.
"""
with smart_open(compressed_file, 'r') as f:
for i, line in enumerate(f):
# Process line by line, decompression happens on-the-fly
if i % 4 == 0: # Header line
print(f"Processing: {line.strip()}")
if i >= 15: # Just demo first 4 records
break
# smartopen works with any compression format automatically
def process_any_format(filepath):
"""Works with .gz, .bz2, .xz, .zst - just works!"""
with smart_open(filepath, 'r') as f:
for record in f:
# Compression is transparent to your code
yield record.strip()
Memory savings:
- Format-agnostic: Automatically detects gzip, bzip2, xz, zstd, and more
- Streaming decompression: Only decompress what you're actively reading
- No intermediate files: Saves disk space equal to uncompressed file size
- No surprises: Same API regardless of compression type
Performance Comparison
Let's quantify the difference with concrete numbers:
import time
import psutil
import os
def monitor_memory(func, *args, **kwargs):
"""Decorator to measure peak memory usage."""
process = psutil.Process(os.getpid())
mem_before = process.memory_info().rss / 1024 / 1024 # MB
start = time.time()
result = func(*args, **kwargs)
elapsed = time.time() - start
mem_after = process.memory_info().rss / 1024 / 1024 # MB
peak_mem = mem_after - mem_before
print(f"Function: {func.__name__}")
print(f" Time: {elapsed:.2f}s")
print(f" Peak Memory Delta: {peak_mem:.1f}MB")
return result
# Hypothetical results with 1GB FASTQ file:
#
# load_into_memory: Time: 45s, Peak Memory: 1200MB ❌
# stream_approach: Time: 32s, Peak Memory: 2MB ✅
# chunked_approach: Time: 38s, Peak Memory: 150MB ✅
For time tracking use funcy.
from funcy import tap, print_durations
import time
def stream_approach():
"""Simulated streaming approach."""
time.sleep(2)
return {"records": 1000000}
def chunked_approach():
"""Simulated chunked approach."""
time.sleep(2.3)
return {"records": 1000000}
@print_durations
def timed_stream():
return stream_approach()
@print_durations
def timed_chunked():
return chunked_approach()
# Run and get automatic timing output
result1 = timed_stream() # Prints: <function timed_stream at 0x...> took 2.00s
result2 = timed_chunked() # Prints: <function timed_chunked at 0x...> took 2.30s
Best Practices Checklist
- ✅ Always stream: Unless you have a very specific reason to load everything, use streaming or chunked approaches. You never know when your file will 200x in size!
- ✅ Use proven libraries: BioPython, pySAM, pandas with
chunksize, polars with streaming - ✅ Monitor memory: Use
psutilormemory_profilerto verify your approach - ✅ Profile your code: Some operations you think are expensive are actually fast, and vice versa
- ✅ Test with real data: Your 1MB test file behaves very differently than a 100GB production file
- ✅ Plan for growth: Design your system assuming files will get out of hand.
Common Patterns by File Type
FASTQ/FASTA
import pysam
with pysam.FastqFile("file.fastq") as fq:
for record in fq:
# record.name: read name
# record.sequence: DNA sequence
# record.query_qualities: quality scores
print(f"{record.name}: {record.sequence}")
BAM/SAM
import pysam
with pysam.AlignmentFile("file.bam", "rb") as f:
for read in f:
# Process read
VCF
import vcf
reader = vcf.Reader(filename="file.vcf.gz")
for record in reader:
# Process variant
CSV/Tabular
from pyarrow import csv
df = csv.read_csv("large_file.csv")
# Process chunk
References
- pySAM FASTQ Files Documentation
- smart_open: Transparent file handling
- BioPython SeqIO
- PyVCF: Variant Call Format handling
Key Takeaway
Large file handling isn't about fancy algorithms—it's about respecting memory constraints and processing data efficiently. The pattern is always the same:
Don't load everything. Stream or chunk instead.
This simple principle will make your bioinformatics software scale from laptop to HPC cluster without modification. Your future self (and your system administrator) will thank you.
Next in the "Do This Not That" series: Parallelization patterns for bioinformatics workflows.