ML/AIOps for Drug Discovery with Boltz2 and MLflow

Overview
If you've ever worked with scientific software you know that it doesn't play well with the other children on the playground. Some of my favorite work related activities are getting rid of code and finding existing code, that I didn't write, to use. MLOps for drug discovery has been an especially challenging arena.
Modern drug discovery relies on complex ML models to virtually screen millions of structures Computational structure prediction is meant to identify promising compounds and understand binding mechanisms.
This, of course, assumes we can:
- Inventory our models
- Run our inference pipelines
- Track our results
- Track additional assets (graphs, plots, images, interim files.)
Much easier said than done!
I've seen MLOps for drug discovery handled all kinds of ways, but the combination of MLFlow has to be my favorite. MLFlow can be run locally. For testing it's backed by a SQLite DB, but can also handle tracking multiple users over multiple experiments. Its an application that I don't have to manage! It also provides a UI for viewing results.
Boltz2, the latest protein-ligand structure prediction model, combined with MLflow's models-from-code framework, creates a powerful MLOps pipeline for tracking, versioning, and deploying AI-driven drug discovery workflows at scale.
Why Models-From-Code for Drug Discovery?
Scientific software is often in it's own category. Custom programs that don't play well with others. As much as I would like to coopt other people's code, it's just not always possible. Today though, it was possible!
MLflow's models-from-code stores your prediction pipeline as readable Python scripts instead of binary artifacts, enabling:
- Reproducibility: Exact code used for each prediction is versioned and auditable
- Transparency: Audit model behavior without unpickling binaries. Version models, metrics, and any other artifacts we want
- Collaboration: Share prediction workflows across research teams without environment conflicts
- Deployment: Load any historical prediction configuration and reuse it with new compounds
Environment Setup
Start by creating a conda environment with all necessary dependencies:
name: boltz-mlflow
channels:
- conda-forge
- defaults
dependencies:
- python=3.12
- pip
- ipython
- pip:
- mlflow
- typer==0.23.1
- click
- boltz[cuda12] @ git+https://github.com/jwohlwend/boltz.git@v2.2.1
# boltz dependencies
- "torch>=2.2"
- "numpy>=1.26,<2.0"
- "hydra-core==1.3.2"
- "pytorch-lightning==2.5.0"
- "rdkit>=2024.3.2"
- "dm-tree==0.1.8"
- "requests==2.32.3"
- "pandas>=2.2.2"
- "types-requests"
- "einops==0.8.0"
- "einx==0.3.0"
- "fairscale==0.4.13"
- "mashumaro==3.14"
- "modelcif==1.2"
- "wandb==0.18.7"
- "click==8.1.7"
- "pyyaml==6.0.2"
- "biopython==1.84"
- "scipy==1.13.1"
- "numba==0.61.0"
- "gemmi==0.6.5"
- "scikit-learn==1.6.1"
- "chembl_structure_pipeline==1.2.2"
# cuda deps
- "cuequivariance_ops_cu12>=0.5.0"
- "cuequivariance_ops_torch_cu12>=0.5.0"
- "cuequivariance_torch>=0.5.0"
Create and activate the environment:
conda env create -f conda_env.yaml
conda activate boltz-mlflow
Implementation
1. MLflow Model Wrapper
The Boltz2Model class wraps Boltz2 predictions in a format that MLflow understands. This enables seamless tracking, versioning, and deployment:
import os
from pathlib import Path
from typing import Any, Dict, Optional
import mlflow
from mlflow.pyfunc import PythonModel
from mlflow.models import set_model
class Boltz2Model(PythonModel):
"""MLflow wrapper for Boltz2 predictions."""
def load_context(self, context):
"""Load model artifacts."""
import torch
from boltz.model.models.boltz2 import Boltz2
checkpoint_path = context.artifacts.get("checkpoint")
if checkpoint_path:
self.checkpoint = torch.load(checkpoint_path, map_location="cpu")
else:
self.checkpoint = None
def predict(self, context, model_input: Dict[str, Any], params: Optional[Dict[str, Any]] = None) -> Dict[str, Any]:
"""Run Boltz2 prediction."""
from boltz.main import predict as boltz_predict
input_path = model_input["input_path"]
out_dir = model_input.get("out_dir", "./predictions")
params = params or {}
boltz_predict(
data=input_path,
out_dir=out_dir,
cache=params.get("cache", "~/.boltz"),
checkpoint=params.get("checkpoint"),
affinity_checkpoint=params.get("affinity_checkpoint"),
recycling_steps=params.get("recycling_steps", 3),
sampling_steps=params.get("sampling_steps", 200),
diffusion_samples=params.get("diffusion_samples", 1),
use_msa_server=params.get("use_msa_server", False),
model=params.get("model", "boltz2"),
)
return {"output_dir": out_dir, "status": "completed"}
set_model(Boltz2Model())
This wrapper:
- Loads context using Boltz to manage the model
- Accepts structured input (input YAML path, output directory)
- Supports parameter overrides for experimentation (sampling steps, recycling iterations, etc.)
- Returns structured output for downstream pipeline steps
2. Experiment Tracking Script
This script orchestrates the complete MLflow workflow:
"""Track Boltz2 predictions with MLflow models-from-code."""
import mlflow
from pathlib import Path
from typing import Dict, Any, Optional
def log_boltz2_run(
input_yaml: str,
experiment_name: str = "Boltz2-Drug-Discovery",
run_name: Optional[str] = None,
tags: Optional[Dict[str, str]] = None,
params: Optional[Dict[str, Any]] = None,
):
"""Log a Boltz2 run to MLflow."""
mlflow.set_experiment(experiment_name)
with mlflow.start_run(run_name=run_name, tags=tags) as run:
# Log parameters
default_params = {
"model": "boltz2",
"recycling_steps": 3,
"sampling_steps": 200,
"diffusion_samples": 1,
"use_msa_server": False,
}
if params:
default_params.update(params)
mlflow.log_params(default_params)
mlflow.log_param("input_file", input_yaml)
# Log the model using models-from-code
model_info = mlflow.pyfunc.log_model(
python_model="boltz2_mlflow_model.py",
artifact_path="model",
input_example={"input_path": input_yaml, "out_dir": "./predictions"},
code_paths=["./code/boltz/src"],
)
# Log input file as artifact
mlflow.log_artifact(input_yaml, "inputs")
return run.info.run_id, model_info
3. CLI Application
For production workflows, a command-line interface provides easier integration with HPC schedulers. Most often virtual screening projects involve screening millions of compounds.
#!/usr/bin/env python3
"""Boltz MLflow CLI - End-to-end experiment tracking and model registration."""
from typing import Optional
import typer
import mlflow
from mlflow.exceptions import MlflowException
app = typer.Typer(help="Boltz structure prediction with MLflow tracking")
@app.command()
def predict(
input_yaml: str = typer.Argument(..., help="Input YAML file for prediction"),
out_dir: str = typer.Option("./predictions", help="Output directory"),
experiment_name: str = typer.Option("boltz-predictions", help="MLflow experiment name"),
model_name: str = typer.Option("boltz-structure-predictor", help="Registered model name"),
register: bool = typer.Option(True, help="Register model after prediction"),
recycling_steps: int = typer.Option(3, help="Number of recycling steps"),
sampling_steps: int = typer.Option(200, help="Number of sampling steps"),
diffusion_samples: int = typer.Option(1, help="Number of diffusion samples"),
):
"""Run Boltz prediction with full MLflow tracking."""
from boltz.main import predict as boltz_predict
from pathlib import Path
import platform
input_yaml = Path(input_yaml)
out_dir = Path(out_dir)
mlflow.set_experiment(experiment_name)
with mlflow.start_run(run_name=f"predict_{input_yaml.stem}") as run:
# Log parameters
params = {
"model": "boltz2",
"recycling_steps": recycling_steps,
"sampling_steps": sampling_steps,
"diffusion_samples": diffusion_samples,
"accelerator": "cpu" if platform.system() == "Darwin" else "gpu",
"input_file": str(input_yaml),
}
mlflow.log_params(params)
mlflow.set_tag("task", "inference")
# Log input
mlflow.log_artifact(str(input_yaml), "inputs")
# Run prediction
typer.echo(f"Running prediction on {input_yaml}...")
boltz_predict.callback(
data=str(input_yaml),
out_dir=str(out_dir),
cache="~/.boltz",
recycling_steps=recycling_steps,
sampling_steps=sampling_steps,
diffusion_samples=diffusion_samples,
use_msa_server=False,
model="boltz2",
accelerator=params["accelerator"],
devices=1,
num_workers=2,
override=True,
seed=None,
msa_server_url="https://api.colabfold.com",
msa_pairing_strategy="greedy",
)
# Log outputs
if out_dir.exists():
mlflow.log_artifacts(str(out_dir), "predictions")
# Log metrics from JSON files
for json_file in out_dir.rglob("*.json"):
try:
import json
with open(json_file) as f:
data = json.load(f)
for key, value in data.items():
if isinstance(value, (int, float)):
mlflow.log_metric(key, value)
except Exception as e:
typer.echo(f"Could not parse {json_file}: {e}")
typer.echo(f"✓ Run ID: {run.info.run_id}")
typer.echo(f"✓ Predictions: {out_dir}")
if __name__ == "__main__":
app()
MLflow Experiment Tracking
Once your models and pipelines are defined, tracking experiments becomes straightforward:
# Track a structure prediction run
run_id, model_info = log_boltz2_run(
input_yaml="./code/boltz/examples/ligand.yaml",
run_name="ligand_binding_screen",
tags={"target": "kinase", "stage": "hit_discovery"},
params={"sampling_steps": 200, "use_msa_server": True}
)
# Load and reuse the tracked model
loaded_model = mlflow.pyfunc.load_model(model_info.model_uri)
result = loaded_model.predict(
{"input_path": "new_ligand.yaml", "out_dir": "./new_predictions"},
params={"recycling_steps": 5}
)
Visualizing Experiments
MLflow's web UI provides comprehensive experiment tracking and comparison. To access it remotely:
mlflow ui \
--host 0.0.0.0 \
--port 5000 \
--allowed-hosts "*" \
--cors-allowed-origins "*"
Then navigate to http://<your-server>:5000 to view runs, compare parameters and metrics:
The runs tab shows all tracked experiments with their associated parameters and outputs:
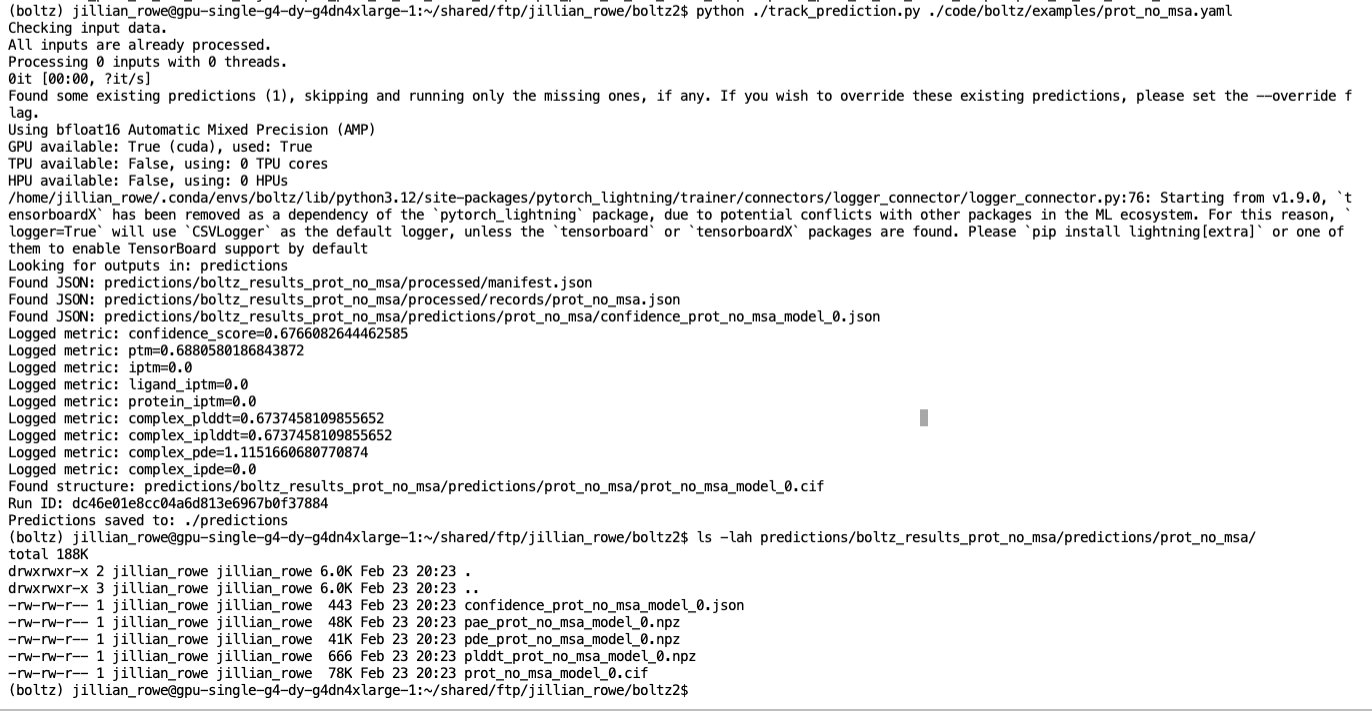
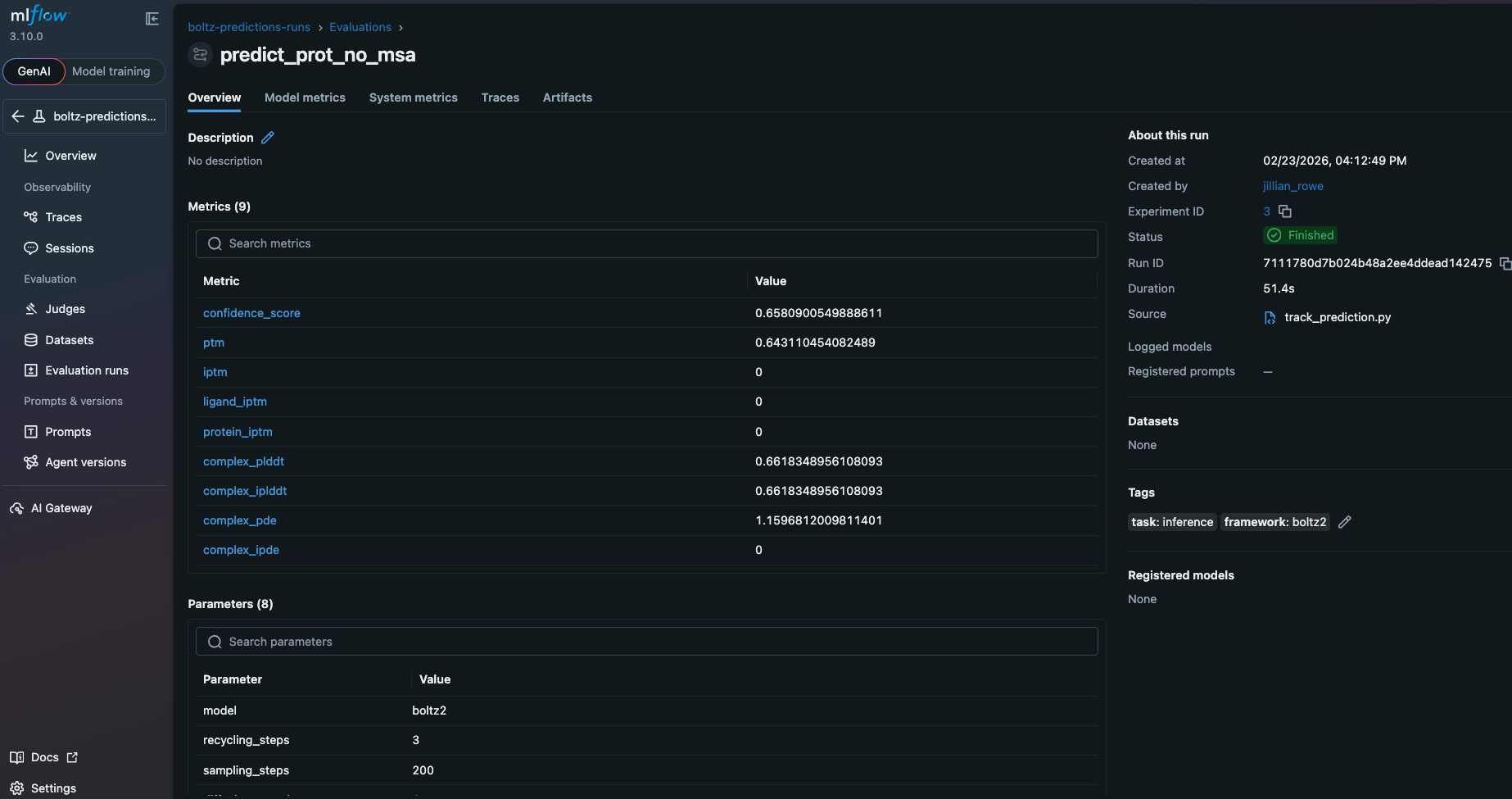
The metrics section displays computed properties from predictions:
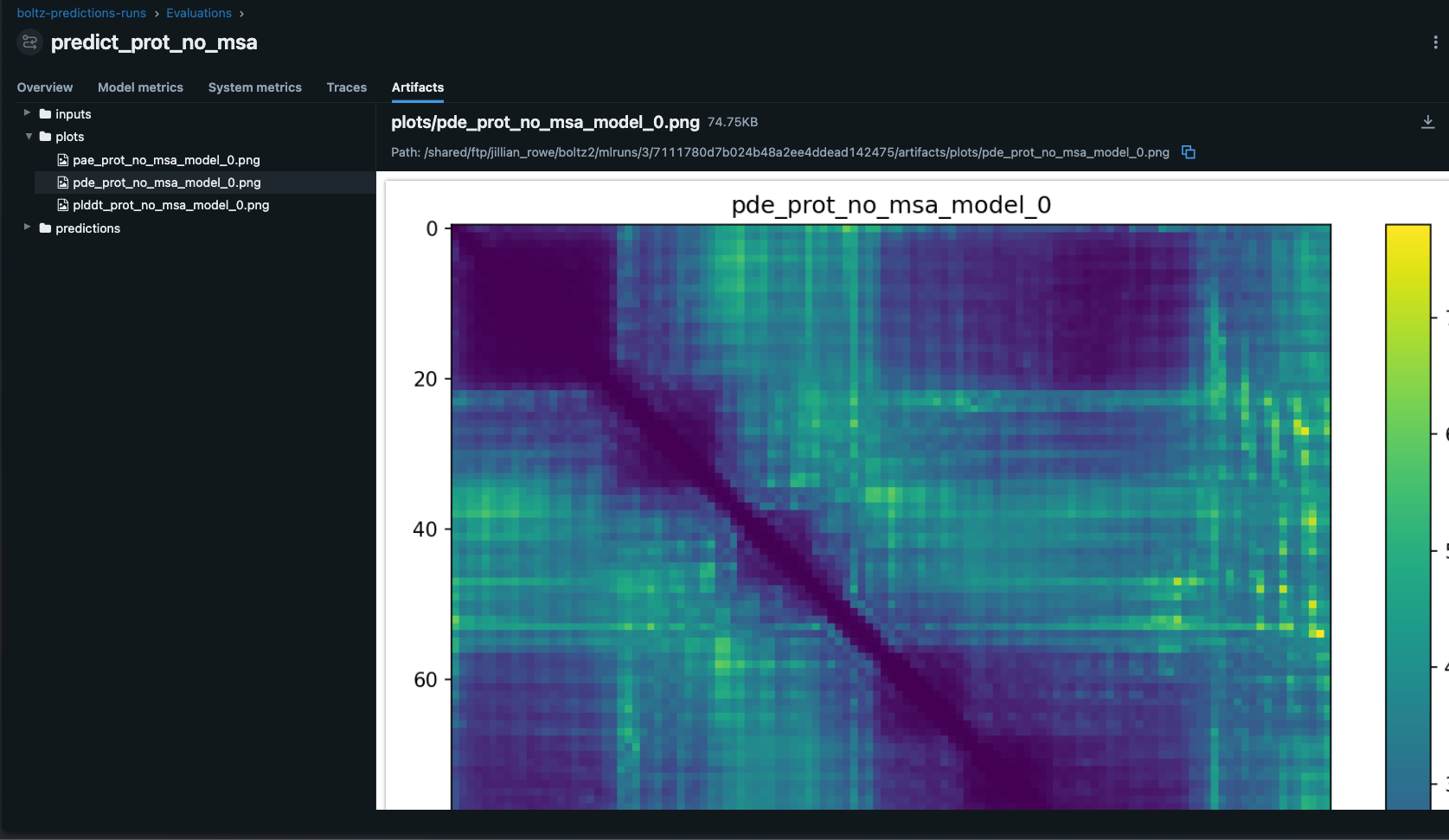
The artifacts section stores input files, output structures, and prediction confidence data:
Best Practices for Drug Discovery MLOps
1. Experiment Organization
Structure experiments by discovery stage:
- Hit-Discovery: Initial screening with binary affinity predictions (
affinity_probability_binary). Use lower sampling steps for throughput - Hit-to-Lead: Optimization using continuous affinity predictions (
affinity_pred_value). Increase sampling for accuracy - Lead-Optimization: Fine-tuning with increased sampling steps (400+) and multiple diffusion samples
Tag all runs with compound ID and discovery stage for easy querying and filtering.
2. Parameter Tracking
In virtual screening we always log:
- Sampling parameters (steps, samples, recycling iterations)
- MSA configuration (server usage, pairing strategy)
- Target information (protein, ligand, binding site)
- Computational resources (GPU type, memory used, runtime)
- Input file hashes for full traceability
3. Artifact Management
Store:
- Input YAML files (enables full reproducibility)
- Generated structures (PDB/mmCIF format)
- Confidence metrics (pLDDT, PAE matrices)
- Affinity predictions and probability scores
- MSA data (if custom, for complex targets)
4. Security
Never hardcode credentials in scripts. Use environment variables:
# Use environment variables
os.environ["BOLTZ_MSA_USERNAME"] = "user"
os.environ["BOLTZ_MSA_PASSWORD"] = "pass"
Or load from secure credential managers in your HPC environment.
5. Reproducibility
Tag runs with:
- Compound IDs
- Target names
- Discovery stage
- Date/batch information
- User/team information
This enables filtering, comparison, and reconstruction of any discovery campaign.
Advanced: Batch Screening Campaigns
For high-throughput screening across multiple compounds:
def track_screening_campaign(ligand_dir: Path, target: str):
"""Track multiple ligand predictions across a target."""
mlflow.set_experiment(f"Screening-{target}")
for yaml_file in ligand_dir.glob("*.yaml"):
with mlflow.start_run(run_name=yaml_file.stem):
mlflow.log_params({
"target": target,
"ligand": yaml_file.stem,
"model": "boltz2",
})
model_info = mlflow.pyfunc.log_model(
python_model="boltz2_mlflow_model.py",
artifact_path="model",
input_example={"input_path": str(yaml_file)},
code_paths=["./code/boltz/src"],
)
mlflow.log_artifact(str(yaml_file), "inputs")
# Load and run prediction
loaded_model = mlflow.pyfunc.load_model(model_info.model_uri)
out_dir = Path("./screening_results") / yaml_file.stem
out_dir.mkdir(parents=True, exist_ok=True)
result = loaded_model.predict(
{"input_path": str(yaml_file), "out_dir": str(out_dir)}
)
mlflow.log_artifacts(str(out_dir), "predictions")
Integration with HPC Schedulers
Since we created a CLI submitting to clusters is trivial:
#!/bin/bash
#SBATCH --job-name=boltz-screening
#SBATCH --nodes=1
#SBATCH --gpus-per-node=1
#SBATCH --time=04:00:00
source activate boltz-mlflow
python boltz_mlflow_cli.py predict \
input_ligands.yaml \
--out-dir ./predictions \
--experiment-name "HT-Screening-2026-04" \
--sampling-steps 200 \
--recycling-steps 3
In a normal screening campaign you would probably have the input_ligands.yaml read in as an array of files, with one ligand per HPC / Batch / Nextflow job.
Key Advantages
- Transparent Tracking: All prediction code visible in MLflow UI for full auditability
- Version Control: Track model code changes alongside predictions—critical for regulatory submissions
- Deployment Ready: Load any historical prediction configuration and apply to new compounds
- Reproducibility: Exact computational conditions captured for every prediction
- Scalability: Trivially extends to high-throughput screening campaigns
References
Conclusion
Combining Boltz2's cutting-edge structure prediction capabilities with MLflow's models-from-code framework creates a robust, auditable ML/AIOps pipeline for drug discovery. By systematically tracking experiments, parameters, and artifacts, you gain the reproducibility needed. All without having to orchestrate your own MLOps system!
The code-based approach to model versioning eliminates black-box artifacts, enables seamless collaboration across teams, and provides the transparency that research teams demand. Start small with individual prediction tracking, then scale to high-throughput screening campaigns.